|
|
|
| CHARCOT MARIE TOOTH HASTALIĞI NEDİR ? | ||||
|
CMT, ilk kez 1886 yılında Fransız Jean Martin Charcot ve Pierre Marie, İngiliz Howard Henry Tooth adında üç ayrı hekim tarafından tanımlanmış nörolojik bir hastalıktır. Adı duyulmamış olsa da oldukça yaygındır. Ancak diğer nörolojik hastalıkların tersine CMT beyni etkilemez, hayati tehlike yaratmaz. Periferal sinirlerde, beyin ve omurilik ile kaslar ve duyu organları arasındaki bağlantıyı sağlayan sinir hücreleri kollarında hasara neden olur. Bu sinir hasarı -veya nöropati- ayak, bacak, el ve kollarda kas gücünün zayıflamasına, kas kitlesinin azalmasına, duyu kaybına yol açar. CMT uzun süreyle bazı kimyasal maddelerin etkisi altında kalınmaktan kaynaklanan çıkan sinir hasarı olan edinilmiş nöropatiye benzerlik gösterir. CMT bulaşıcı değildir, kalıtımla geçer. Bu özellikleri nedeniyle CMT ye "kalıtsal, hareket ve duyu sinirleri nöropatisi" (HMSN) de denir. İlk olarak alt bacaktaki peroneal kas zayıflamaya başladığından, bazı hekimler CMT ye peroneal muskuler atrofi adını vermektedir. |
||||
|
||||
|
Herbiri, şiddet, başlama yaşı, ilerlemesi ve belirtileri açısından birbirinden farklı formlarda göründüğü için CMT ye pek çok isim verilmiştir. Örneğin Dejerine-Sottas hastalığı CMT nin çocukluk dönemde görülen şiddetli bir formudur. CMT nin kesin bir tedavisi olmamakla birlikte, bazı |
||||
| CMT nin Sebepleri | ||||
|
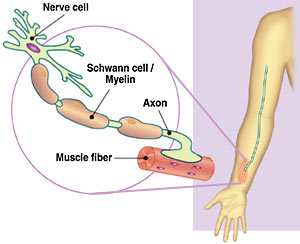
CMT gendeki bozulmadan kaynaklanır. Genler vücutta gerekli fonksiyonları yerine getiren proteinleri üreten yerlerdir. Her CMT formu spesifik bir gene bağlıdır ve bu genlerin tümü periferal sinirler için protein üretir. Periferal sinirler beyin ile vücut arasındaki iletimi sağlarlar. Örneğin bacağınızı hareket ettirmek istediğinizde beyin, omurilikteki kas kontrol edici sinir hücrelerine elektriksel sinyal gönderir sonra bu hücreler sinyali bacak kaslarındaki periferal sinirlere aktarırlar. Bacağınız incindiğinde acı duyarsınız çünkü acıya duyarlı sinir hücreleri periferal sinirler vasıtasıyla beyine elektrik sinyali yollamıştır. Periferal sinirler akson denen uzantılardan oluşur, aksonlar duyu hücrelerinden kas kontrol edici sinir hücresine doğru uzanır ve omurilikten gelen elektrik sinyallerini taşır. Hareket etmek ve hızla reaksiyon gösterebilmek için aksonlar sinyali saniyeden çok küçük bir zaman dilimi içinde taşırlar. Aksonların performansının artması için herbiri myelin denen bir kılıfla kaplanmıştır. Elektrik tellerinin plastik kaplama ile izole edilmesi gibi myelin de aksonlardaki elektrik sinyallerini izole eder ve aynı zamanda aksonun beslenmesini sağlar. CMTde, her biri hastalığın spesifik bir tipi ile bağlantılı bir düzineden fazla gen devreye girmektedir. Bu genlerin bir kısmı aksonlarda , diğer bir kısmı ise myelinde ihtiyaç duyulan proteini üretmektedir. Defektif akson genleri akson fonksiyonlarında zayıflamaya neden olurken, defektif myelin genleri de myelin çökmesine sebep olur. Her iki durumda da sonuç aynıdır :akson veya myelindeki defektler aksonlarda ilerleyici hasara neden olur. Vücuttaki en uzun aksonlar hasara karşı özellikle duyarlı olduğundan, CMT en çok el ve ayaklarda hareket ve duyu problemi yaratmaktadır. |
||||
|
Kas Zayıflaması Genel olarak CMT hastalarında yavaş ilerleyen kas gücü zayıflaması, el-kol-ayak-bacak kaslarında kitle azalması görülür. Baş ve boyuna yakın kaslarda zayıflama görülmez. Kas zayıflaması çoğunlukla ayak ve ayak bileğinde başlar ve düşük ayak şeklinde kendini gösterir, hasta ayağını bilekten yukarı kaldıramaz ve ayak parmakları yürürken aşağı doğru kıvrılır. Düşük ayak sık sık tökezlenmeye neden olur ve zayıflama arttıkça hastanın yürüyüşü anormal bir hal alır. Pek çok CMT hastası, düşük ayağın neden olduğu sıkça tökezleyip düştüklerini farkettikten, bilek burkulma ve kırıklarıyla karşılaştıktan sonra bir nörologa gitmiştir. Bu problemler ortaya çıkarken bazıları da bileği destekleyen bot veya yüksek tabanlı ayakkabı giyerek sorunun üstesinden gelebileceğini düşünür. Bir diğer hasta grubu ayağı ve bileği sıkıca saran, kolayca çıkarılabilir aya-bilek ortezi gibi bacak ortezi kullanmak ister. Bir ara özel ayakkabı gerektiren metal destekler de kullanılmıştır ancak şimdi kullanan kişinin bacağına tam olarak oturan ve pantolonun altına tenis pabuçlarıyla giyilebilen ortezler hafif plastik maddeden yapılmaktadır. Daha çok proksimal kasları zayıflamış kişiler için bacağını dizden yukarı kaldırabileceği "ayak-bilek-diz" ortezi geliştirilmiştir. Bazı ortezler hareketi engelleyip daha çok destek sağlarken bazıları da bilek ve diz hareketine izin verir. İlerlemiş veya şiddetli CMT hastaları elektrik motorlu scooter veya tekerlekli sandalye ile çevrede dolaşma imkanı bulabilir. CMT hastalarında solunum kaslarının zayıflaması çok nadir görülür fakat meydana geldiğinde hayati tehlike yaratabilir. Sık ve kısa nefes alıyorsanız mutlaka uzman bir hekim tarafından solunum kontrolü yapılmalı ve geceleri ventilatör veya BPAP kullanılmalıdır. Bazı CMT hastalarında görülen tremor genelde engel yaratmaz ve hastaya rahatsızlık vermez. Belirgin tremor görülen CMT ye Roussy-Levy Sendromu adı verilir. |
||||
| Kontraktür ve kemik deformiteleri | ||||
|
Pek çok CMT hastasında sonunda ayak ve ellerde deformite ile sonuçlanan eklem sertleşme ve daralmaları görülür. Eklem etrafındaki bir kısım kaslar zayıflarken bazılarının gücünü koruması yüzünden kontraktür kaçınılmazdır. Zamanla eklem yakınındaki kemikler de anormal pozisyon sergiler. Örneğin ayağı bilekten yukarı kaldıran kas zayıfladığında ayağı aşağı indiren ve yere doğru kıvıran kasın yarattığı kontrakt, kısa ayak ve yüksek köprü ile en yaygın görülen ayak deformitesine neden olur. Kontraktür kötüleştiği zaman ayak parmakları fleksiyon pozisyonunda dondurulabilir. CMT hastalarının küçük bir bölümünde kas zayıflamasının farklı bir görünümü olan "düz-taban" gelişir. Yürüyüş sırasında bu deformiteler ağrılı sürtünme, su dolu kabarcıklar ve nasır oluşumuna yol açabilen parmak,topuk ve bilek üzerinde beklenmedik sürtünmelere neden olur. Tedavi edilmezse kontraktür ve sekonder kabarcıklar zamanla kötüleşir ve giderek yürümek güçleşir. CMT gelişimi içinde, eldeki kontraktürler parmakları fleksiyon pozisyonunda sabitleyebilir, nadir vakalarda şiddetli proksimal zayıflama skolyoz ve kifoza neden olabilir. Yine şiddetli CMT hastalarının küçük bir bölümünde erken yaşlarda bel kayması gelişebilir. Kasların incelmesine ve kontraktur oluşumuna engel olmanın en etkili yolu düşük etkili egzersiz ve gerilme hareketlerinden oluşan düzenli fizyoterapi programı uygulamaktır. |
||||
| Duyu Kaybı ve Semptomları | ||||
|
Duysal aksonlarda tahribat oluştuğundan CMT hastalarının ayak bacaklarında ısı, dokunma ve ağrıya karşı duyarlık azalır, CMT hastaları ayaklarının üşüdüğünden şikayetçi olurlarsa da, duysal kayıpların çoğu nörolojik bir inceleme ile tespit edilemez. Ancak yine de bu şikayeti ciddiye almak gerekir. Ayak deformitelerinin neden olduğu düzenli yıpranma beraberinde ağrı duyusunun yokluğu CMT hastalarında farkına varılmadığı taktirde şiddetli enfeksiyonların oluşabileceği ülserasyon riskini yaratır. Ayak deformitesi olan CMT hastası düzenli olarak yara kontrolünden geçmelidir. Tam tersine bazı CMT hastalarında ağrılı kas krampı ve nöropatik ağrı karışımı şiddetli ağrı görülebilmektedir. Bu ağrının dış tetikleyicisi olmamakla beraber aksonlardaki defektif sinyaller olabileceği düşünülmektedir. Her ağrı tipi uygun bir medikasyonla yatıştırılabilir. Nadir vakalarda duysal kayıp içinde işitme kaybı ve bazen sağırlık ta görülebilmektedir. Muhtemel problemleri gözlemlemekle gerektiğinde uygun tedavi yöntemi arama imkanı elde edilir. |
||||
| Zararlı İlaçlar | ||||
|
Bazı ilaçlar ve aşırı alkol edinilmiş nöropatiye yol açmakta ve CMT yi alevlendirmektedir.Vaka çalışmaları kemoterapi ilaçlarından vincristine in CMT li hastalarda hızlı kötüleşmeye neden olduğunu göstermiştir. Bilacın ilk kez kullanımından önce CMT üzerindeki olası etkileri yönünden doktora danışılması yerinde olur. |
||||
| CMT Tipleri Nelerdir? | ||||
|
Birbirinden farklı pek çok CMT tipi başlangıç yaşı, kalıtım paterni,şiddeti ve akson veya myelin defektine bağlı oluşu ile birbirinden ayırt edilebilir. Bu ayırımlar yararlıdır ayrıca çok sayıda genetik defekt CMT ye yol açtığından bazılarının iki farklı CMT tipinin sınırında ve diğer bazı kişilerin de spesifik alt tipler olduğunun farkında olmak çok önemlidir. |
||||
| CMT1 ve CMT2 | ||||
| Başlama yaşı :
Genellikle çocukluk ve erginlik Kalıtım- otozomal dominant Özellikler-CMT nin en yaygın 2 formudur. (aslında CMT 1 in, kromozom 17 deki PMP22 geni defektinin neden olduğu alttipi CMT1A vakaların yaklaşık %60 nı teşkil eder.) CMT1 in nedeni demyelinasyon, CMT2 nin aksonopatidir ancak her ikisi de klasik semptomları gösterir. CMT2 bazen tedavisi mümkün olan "restless bacak sendromu" ile ilişkilidir. |
||||
| CMT3 ve CMT4 | ||||
|
Başlama-Çocuk
yaşlar |
||||
| CMTX | ||||
|
Başlangıç-Çocukluk veya erginlik Kalıtım-X-bağlantılı Özellikler-CMTX CMT1 ve CMT2 ile benzer semptomları taşır fakat çoğunlukla erkeklerde kadınlara oranla daha şiddetlidir. |
||||
| Konjenital Hypomyelinasyon Nöropatisi (CHN) | ||||
| Başlangıç-Doğuştan
veya doğuştan hemen sonra Kalıtım-Otozomal resesif, spontan Özellikler-Diğer CMT tiplerinin tersine CHN mevcut myelinin çökmesinden ziyade doğuştan myelinin az olmasından kaynaklanır.Genetik ve klinik olarak DS ye benzerse de başlangıcı erken yaşlardadır ve ilerlemesi yavaştır.Baze solunum sorunlarına bağlı ölüm vakalarına rastlanırsa da hastaların çoğu güçlü bir yaşam sürer. |
||||
HNPPBaşlangıcı-çoğunlukla adolesan dönemde |
||||
| CMT Tanısı Nasıl Konur? | ||||
|
Alt bacaktaki zayıflık ve ayak deformitesi CMT nin ilk işaretidir ancak bunlar tanı için yeterli değildir fiziksel muayene ile distal zayıflık ve duyu kaybı olup olmadığı aranır. Bacaktaki güç kaybını test etmek için hasta parmak uçlarında yürütülür, bacağına zıt güç uygulanıp bacağını kaldırması istenir.Duyu kaybını anlamak için tendon refleksi testi yapılır çünkü CMT hastalarında bu refleks azalmıştır veya hiç yoktur. İlk değerlendirmelerde hastanın aile öyküsü alınır.Ailede CMT benzeri fiziksel hareketlerden kaynaklanan sinir hasarı belirtileri beraberindeki semptomların varlığı CMT veya başka bir kalıtsal nöropatiyi işaret eder. Ailede öykünün bulunmaması CMT yi dışarıda bırakmaz ancak diabet, bazı ilaçları uzun süre kullanma ve diğer potansiyel nöropatiler araştırılmalıdır. CMT üzerinde duruluyorsa sonraki aşamada genetik testler yapılır. Alınan kan örneklerinde CMT ye neden olabilecek bilinen genetik defektler aranır. Pozitif test sonuçları doğru tanı koymayı ve aile planlaması için yararlı bilgiler sağlar. Tekrar belirtelim test sonuçlarının negatif çıkması CMT olasılığını uzak tutmamalıdır. Çünkü hastada bilinmeyen bir genetik defektin yol açtığı CMT bulunabilir ve çok nadir olduğu için yapılan test uygun olmayabilir. Aynı zamanda sinirlerce iletilen elektrik sinyallerinin güç ve hızını ölçen sinir iletim hızı testleri uygulanır. Bu test için, EKG cihazında kullanılanlara benzeyen yüzey elektrodunu sinir üstüne gelecek şekilde deride çeşitli noktalara konur bir elektrod sinirde elektriksel cevap üretecek orta şiddette şoklar verirken diğeri sinirin verdiği cevabı kaydeder. (Gerekirse hastanın şoklardan rahatsız olmaması için bir sakinleştirici veya hafif anestezi verilebilir) Gecikmiş cevaplar demyelinasyon küçük cevaplar ise aksonopati işaretidir. Bu nedenle NCV testi CMT1 ve CMT2 yi ayırt etmede çok yararlıdır. Bunun dışında tanı koymak için kastaki elektrik sinyallerini ölçen Elektromyografi çekilir hasta sinirden küçük bir parça alınarak biyopsi yapılır. |
||||
| CMT Ailede Bulaşıcı mıdır? | ||||
| Kendisine genetik bir bozukluğu olduğu anlatılan CMT hastalarının ilk sorusu hastalığının ailede bulunmadığı halde nasıl genetik karakterli olabileceğidir. Açık bir aile öyküsü olmamasına rağmen CMT soydan soya geçebilir. Çünkü, CMT, soyağacında izlenmesi hiç te kolay olmayan birbirinden farklı 3 yolla nesilden nesile aktarılır : x-bağlı geçiş, otozomal dominant ve otozomal resesif. | ||||
| x-bağlantılı geçiş genetik defekt veya mutasyonun x kromozomu üzerinde lokalize olması demektir. 2 adet x kromozomu bulunan kadınlarda bir kromozom üzerindeki normal gen kopyası, defektif kopyanın etkilerini en azından kısmen yok edebilir. O nedenle x-geçişli hastalıklar erkekleri kadınlara oranla daha şiddetli etkiler çünkü erkekte bir x kromozomu vardır. CMTX gibi x-geçişli hastalıklar babadan oğula geçmez. | ||||
| Otozomal-Mutasyon
x veya y dışındaki kromozomda meydana gelmiş demektir. O nedenle otozomal
hastalıklar erkek ve kadını eşit sayıda etkiler. Otozomal resesif ise, hastalığın tam oluşması için 2 adet defektif gen kopyasına gerek var demektir. Bir kopya normal olarak bu hastalığı taşımayan anne-babadan gelmemiş demektir. Otozomal dominant, hastalığın oluşması için defektif genden bir kopyanın bulunmasının yeterli olmasıdır. O taktirde defektif geni kalıtımla alan kişi de ebeveyni gibi hasta olur |
||||
| CMT otozomal dominant olarak aktarılmışsa bunu soyağacından izlemek mümkündür. x-geçişli veya otozomal resesif tip CMT de "out of blue" oluşabilir. Fakat aslında anne veya hem anne hem baba genetik mutasyonun taşıyıcısı olabilir. Bir çok anne-baba çocuklarında hastalık çıkana kadar taşıyıcı olup olmadıklarını bilmez. | ||||
| CMT herhangi bir neslin bir bireyinde meydana gelebilir, çocuğun oluşumu sırasında meydana gelen bir mutasyon spontan mutasyondur ve sonraki kuşağa geçer.Kalıtsal olarak hastalığı alma ve aktarma riski büyük ölçüde CMT nin tipine bağlıdır bunu anlamanın en iyi yolu da genetik ve klinik tahliller yaptırmaktır. | ||||
|
|
||||